第一节 DNA的克隆
以质粒或噬菌体作载体进行DNA克隆,在理论上是很简捷的,即用一种限制酶切割质粒DNA,然后在体外与外源,DNA(如干扰素基因,生长激素基因)相连接,再用所得到的重组质粒转化细菌如大肠杆菌,以鉴定重组DNA。
通常所用的质粒载体有pBR322、pUC18、pUC19等,噬菌载体有M13噬菌体载体等。
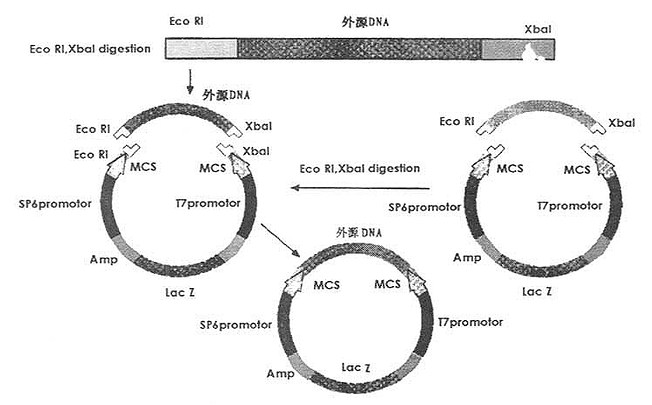
图24-1 质粒载体的定向克隆
一、质粒或噬菌体载体与外源DNA的连接反应
外源DNA片段末端的性质,以及质粒载体和外源DNA上限制酶切位点的性质决定了质粒或噬菌体与外源DNA连接反应的不同策略。
(一)外源DNA带有非互补突出端的片段
用两种不同的限制酶分别消化质粒等载体和外源DNA,可以产生带非互补突出端的片段,这是最容易连接的片段,如图24-1所示。
将已经连接外源DNA的细菌质粒载体转化感受态的大肠杆菌,然后在含有AP等抗生素的平板上鉴定阳性重组体菌落,具体方法有:①检查α互补能力的丧失情况;②对小量制备的质粒DNA进行限制酶切的分析;③核酸杂交。
(二)外源DNA带有相同末端(平端或粘端)的片段
带有相同末端(平端或粘端)的外源DNA片段必须克隆到具有匹配末端的线状质粒或噬菌体载体中,在连接反应中,质粒或噬菌体载体可能发生自身环化,也可能形成串联寡聚物。因而,常常使用碱性磷酸酶去除5’磷酸基团以抑制质粒DNA的自身连接和环化。用细菌碱性磷酸酶(BAP)或牛小肠碱性磷酸酶(CIP)去除线状DNA两端的5’磷酸可以最大限度地减少质粒DNA的自身环化。而具有5’末端磷酸的外源DNA片段可有效地与去磷酸化质粒DNA相连接,产生一个含有两个切口的开环分子。因为环化DNA(即使只带切口的环状DNA)的转化效率比线状DNA高得多,所以大多数转化体都含有重组质粒,如图24-2所示。
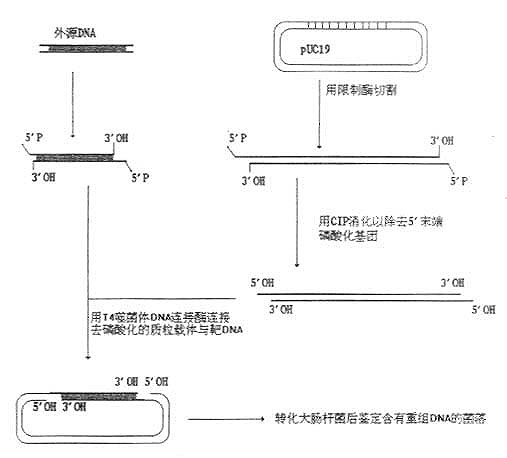
图24-2 利用磷酸酶防止载体DNA的重新环化
(三)外源DNA带有平端的片段
外源DNA片段带有平端的片段,平端的连接效率比带有互补末端的DNA要低得多。因此,涉及平端分子的连接反应所要求的T4噬菌体DNA连接酶的浓度和外源及质粒DNA或噬菌体DNA的浓度都要高得多。
(四)连接反应的体系
10×连接缓冲液1μl
10mMATp 1μl
质粒或噬菌体DNa200ng~1μg
外源DNa 200ng~1μg
T4噬菌DNA连接酶0.5~2nuits
4-6℃温育8~24h
连接反应结束时,取1~5μl上述反应混合物转化200μl的大肠杆菌感受态细胞,即可将外源DNA与载体接连起来。
二、大肠杆菌感受态细胞的制备和转化
细菌转化的方法多以Mendel和Higa(1970)的发现为基础,其基本方法是用冰预冷的CaCl2或多种2价阳离子等处理细菌,使之进入感受态得以转化。
用CaCl2制备新鲜或冷冻的大肠杆菌感受态细胞,常用于成批制备感受态细菌。本法适用于大多数大肠杆菌菌株,且迅速、重复性好。操作过程简述如下。
1.从37℃培养16~20h的新鲜平板中挑取一个单菌落(如大肠杆菌DH52),或1ml新鲜的16~20h过夜培养物,转到一个含有100mlLB培养基的1L或500ml烧瓶中。于37℃剧烈振摇培养约2~3h(旋转摇床200~300r/min),每隔20~30min测量OD600值≈0.4。
2.在无菌条件下将细菌转移到一个,用冰预冷的50ml聚丙烯离心管中,在冰上放置10~20min。
3.于4℃用SorvallGS2转头(或与其离心管相配的转头)以4000r/min离心10min,以回收细胞。
4.倒数培养液,将管倒置1min以使最后残留的痕量培养液流尽。
5.以10ml用冰预冷的0.1mMCaCl2重悬每份沉淀,放于冰上。
6.于4℃用SorvallGS3转头(或与其相应的转头)以4000r/min离心10min,以回收细胞。
7.倒出培养液,将管倒置1min以使最后残留的痕量培养液流尽。
8.每50ml初始培养物用2ml冰预冷的0.1M CaCl2重悬每份沉定,此时,可以迅速将细胞分装成小份,液氮中冰冻,-70℃贮存备用。
9.用冷却的无菌吸头从每种感受态细胞悬液中各取200μl转移到无菌的微量离心管中,每管加DNA或连接反应混合物(体积≤10μl,DNA≤50ng),轻轻旋转以混匀内容物,在冰中放置30min。
10.将离心管放到预加温到40℃的循环水浴中的试管架上,放置90s~2min,不要摇动试管。
11.快速将管转移到冰浴中,使细胞冷却1~2min。
12.每离心管加800μlSOC培养基,用水浴将培养基加温到37℃,然后将管转移到37℃摇床上,温育45min使细菌复苏,并且表达质粒编码的抗生素抗性标记基因。
13.将适当体积(每个90mm平板可达200μl)已转化的感受态细胞转移到含200mmol/l MgSO4和相应抗生素的SOB培养基上。
14.将平板置于室温至液体被吸收。
15.倒置平皿,于37℃培养,12~16h后可出现菌落。
三、用M13噬菌体转染感受态细胞
1.感受态细胞的制备
(1)用JM101或TG1等菌的培养菌液在M9基本培养基平板上的划线培养,于37℃温育24~36h。
(2)批量制备冻存的感受态细胞的方法,同大肠杆菌感受态细胞的制备。
2.用M13噬菌体转染感受态细胞
(1)用2×YT或SOB培养液于37℃持续地振摇,将用于铺平板的细胞(JM101或TG1等)培养过夜。
(2)从-70℃以下冰箱中取出一份冻存的JM101或TGI感受态细菌,于室温慢慢融化,立即放在冰上10min。
(3)于各感受态细胞管中,加入连接反应液,应同时做两个对照,一个加5pgM13噬菌体双链环状DNA,另一个则完全没有DNA。轻弹管外壁使其混匀,冰浴30min。
(4)取数支无菌培养管,分别加入3ml熔化的2×YT或SOB顶层琼脂,置47℃以备步骤6使用。
(5)将装有感受态细胞和DNA的培养管放入42℃水浴,温育整整90s,立即将管放回冰浴之中,2min后各管加入1ml过夜培养的JM101或TG1等菌液,混匀。
(6)在步骤(4)准备的装有溶化的2×YT或SOB顶层琼脂的管内各加入40μlX-gal溶液(20mg/ml,溶于二甲基甲酰胺)和4μlIPTG溶液(200mg/ml)振荡混匀,分别取(5)中混合物各300μl加入各管,振荡混匀,立即将管内混合物倾入标记好的LB琼脂平板上,轻轻旋动平皿,以使细菌与顶层琼脂分布均匀。
(7)盖好平皿,于室温放置5min,使顶层琼脂凝固,将平板倒置于37℃培养。4个h后噬斑开始出现,8~12h后菌斑不再变化。野生型M13噬菌体形成的噬斑为深蓝色,重组噬菌体则可形成无色噬斑。
四、含重组质粒的细胞菌落的鉴定
含重组质粒的细胞菌落常用的鉴定方法有:①小规模制备质粒DNA进行限制酶切分析;②α互补;③插入失活;④杂交筛选。下面简要介绍小规模制备质粒DNA进行限制性酶切分析。小规模制备质粒DNA可用天美等公司的Wizard Minipreps DNA试剂盒或采用下述方法:
1.挑取一些独立的转化菌落进行小规模培养,用无菌牙签或挑种环挑取单菌落于2ml含有相应抗生素的LB液体培养基中,于37℃剧烈振摇下培养过夜。
2.将1.5ml培养物倒入微量离心管中,用微量离心机于4℃以12000g离心30s,将剩余的培养物贮存于4℃。
3.吸取培养液,使细菌沉淀尽可能干燥。
4.将细菌沉淀重悬于200μl溶液Ⅰ中,剧烈振荡。
溶液Ⅰ 25mmol/L Tris・HCl(pH8.0)
10mmol/LEDTA(pH8.0)
溶液Ⅰ可成批配制,每瓶约100ml,高压下101bf/m2(6.895×104pa)蒸气灭菌15min,贮存于4℃。
5.加200μl新配制的溶液Ⅱ。
溶液Ⅱ 0.2mol/L NaOH
1% SDS
盖紧管口,快速颠倒离心管5次,以混合内容物。应确保离心管的整个表面均与溶液Ⅱ接触。不要振荡,将离心管放置于冰上。
6.加200μl溶液Ⅲ
溶液Ⅲ 5mol/l 乙酸甲 60ml
冰乙酸11.5ml
水28.5ml
盖紧管口,将管倒置,温和振荡10min,使溶液Ⅲ在粘稠的细菌裂解物中分散均匀,之后将管置于冰上3~5min。
7.用微量离心管于4℃以12000g离心5min,将上清转移另一离心管中。
8.加等量酚和氯仿,振荡混匀,用微量离心机于4℃以12000g离心2min,将上清转移到另一离心管中。
9.用2倍体积的乙醇于室温沉淀双链DNA,振荡混合,于室温放置2min。
10.用微量离心机于4℃以12000g离心5min。
11.小心吸去上清液,将离管倒置于一张纸巾上,以使所有液体流出。再将附于管壁的液滴除尽。
12.用1ml70%乙醇于4℃洗涤双链DNA沉淀,按步骤1所述方法去掉上清,在空气中使核酸沉淀,干燥10min。
13.用50μl含无DNA酶的胰RNA酶(20μl/ml)的TE重新溶解核酸,振荡,贮存于-20℃。
14.用适当限制性内切酶分析所得DNA。
五、M13噬菌体重组DNA分子导入大肠杆菌
1.感受态细胞的制备
(1)将1ml过夜培养的细菌(如DH52、TG1、TM101)接种于100ml2×YT培养于500ml烧瓶。于37℃刷烈振荡,通常≥200rpm培养的细菌密度约OD550=0.2~0.5(5×107cells/ml),需2~4h。
(2)将培养物放于冰上致冷10min,于4℃离心细菌培养物,10000rpm离心10min。
(3)弃除上清,将细菌悬浮于原始培养体积的一半(约50ml)的50mMCaCl2和10mmTris・HCl(pH8.0)无菌冷冻液体中。
(4)将细菌悬浮放在冰上约5min,然后将悬浮物于4℃10000/rpm离心10min。
(5)弃上清,悬浮细菌于原始培养体积的1/15的50mMCaCl和10mMTris・HCl(pH8.0)无菌冷冻中。此时的细胞是感受态细胞,即可用于转化。200μl分装于无菌的1.5ml微量离心管中,贮存于-80℃冰箱中。
2.转化程序
(1)取出200μl感受态细胞慢慢使其溶化,并立即放于冰上,加入DNA或连接反应混合物,DNA量通常≤50mg。用手指弹打试管10次,使之混匀,然后放在冰上40~45min。
(2)将管放于42℃水浴中2min。
(3)然后每管直接加入1.0ml2×YT培养基,倒置混匀,并于37℃培养8~12h,干浴或水浴,不需振摇。此期间使细菌生长并开始表达抗菌素。
(4)每200μl转化混合物分别于6个2×YT琼脂培养板上补充相应抗生素,并含有X-gal(20mg/ml)、IPTG(200mg/ml)。
(5)铺板使细菌混合物干后,倒转平板放于30~37℃培养箱中18~22h。克隆在此期间应该出现,否则转化不成功。